The Complete Dummy's Genetics Guide for Idiots, Part Three
applications
In part one of this series, we looked at basic genetics concepts such as DNA, inheritance, and cells and cell division, and in part two we examined more difficult ideas, such as dominance and recessivity, sex-linked traits, genetic pathways, development and environmental effects. From here on, I'm going to assume you're comfortable with all of this, so please review if necessary.
So far, we've just considered situations where everything goes to plan, focussing mostly on theoretical concepts. But real life is messy, so let's conclude this series by switching our attention to some practical applications of genetics, by taking a brief look at chromosome variations, genetic mutation, cancer, selective breeding and disease genetics.
By this point, you should be able to visualize what's going on inside our cells at the genetic level. You know from part one that our 30 trillion body cells each carry a genome consisting of a DNA-protein complex, divided into 23 pairs of chromosomes: 22 autosome pairs plus a pair of sex chromosomes. You also know that our sex cells (eggs or sperm) each carry only one chromosome from each pair, with each single chromosome again becoming one member of a pair following fertilization (the fusing of the sperm with the egg cell to make a zygote).
Remember also from part one that, immediately prior to their separation during meiosis, the members of the chromosome pair exchange segments with one another, a mechanism which greatly increases genetic variation amongst offspring. This exchanging of segments is not quite perfect; there are several ways in which things can go wrong, either with the exchanging process or with the subsequent separation.
As we have seen, every chromosome pair will normally separate during metaphase, with one member of each pair moving into one daughter cell, and the other member of the pair moving into the other daughter cell. However, sometimes this separation does not work correctly for a pair of chromosomes, with the result that one daughter cell receives both members of the pair, while the other receives neither.
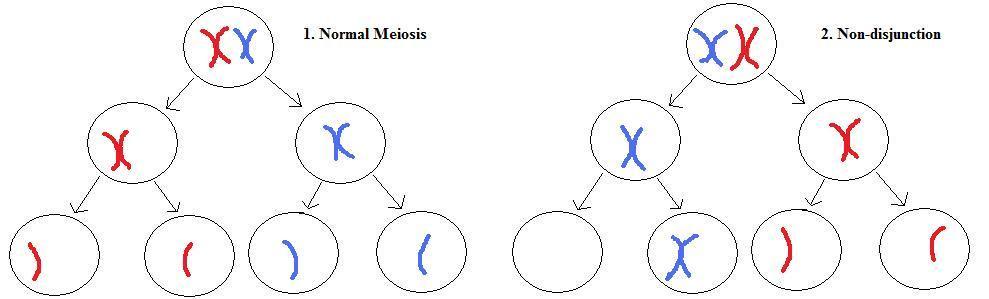 Left: normal meiosis for a single chromosome pair. Top, each chromosome in the pair has duplicated into connected sister chromatids and lined up ready for cell division at metaphase. Middle, the pair separates into separate daughter cells during meiosis I. Bottom, the sister chromatids separate during meiosis II.
Right: non-disjunction meiosis. Normal metaphase (top) and meiosis I (middle), but non-disjunction occurs during meiosis II (bottom). This results in a gamete with a missing chromosome, and another gamete with an extra chromosome. Note that the other chromosomes are omitted for clarity, and that non-disjunction can also occur during meiosis I.
Left: normal meiosis for a single chromosome pair. Top, each chromosome in the pair has duplicated into connected sister chromatids and lined up ready for cell division at metaphase. Middle, the pair separates into separate daughter cells during meiosis I. Bottom, the sister chromatids separate during meiosis II.
Right: non-disjunction meiosis. Normal metaphase (top) and meiosis I (middle), but non-disjunction occurs during meiosis II (bottom). This results in a gamete with a missing chromosome, and another gamete with an extra chromosome. Note that the other chromosomes are omitted for clarity, and that non-disjunction can also occur during meiosis I.
This leads to gametes with an extra or missing chromosome. When a gamete with two copies of a chromosome fuses with a normal gamete with one copy of the chromosome, at fertilization, the resulting zygote will have three copies of that chromosome (a trisomy). When a gamete with zero copies of a chromosome fuses with a normal gamete, the zygote will have only one copy of that chromosome (a monosomy). Although trisomies and monosomies can occur for any chromosome, the only ones compatible with life1 are trisomy 21 (Down Syndrome) and those affecting the sex chromosomes.
An individual with Down Syndrome has three copies of chromosome 21, and two copies of the other chromosomes. This extra genetic material leads to the collection of attributes that are characteristic of the syndrome.
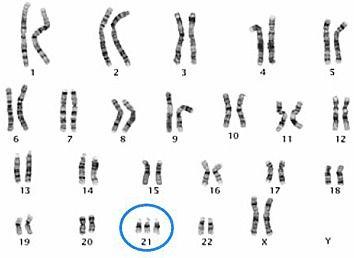 Trisomy 21 results from an extra copy of chromosome 21, as seen in the karyogram of this female.
Trisomy 21 results from an extra copy of chromosome 21, as seen in the karyogram of this female.
Monosomies of the autosomes involve missing genes; all are too severe for a pregnancy to continue for more than a brief period.2 Monosomy of the X-chromosome (XO) is called Turner Syndrome. There is one X-chromosome and no Y-chromosome. The effects of this are usually relatively mild, resulting in females with short stature and infertility but normal intelligence. Klinefelter syndrome (XXY) is another relatively mild condition, resulting in males with an extra X-chromosome.
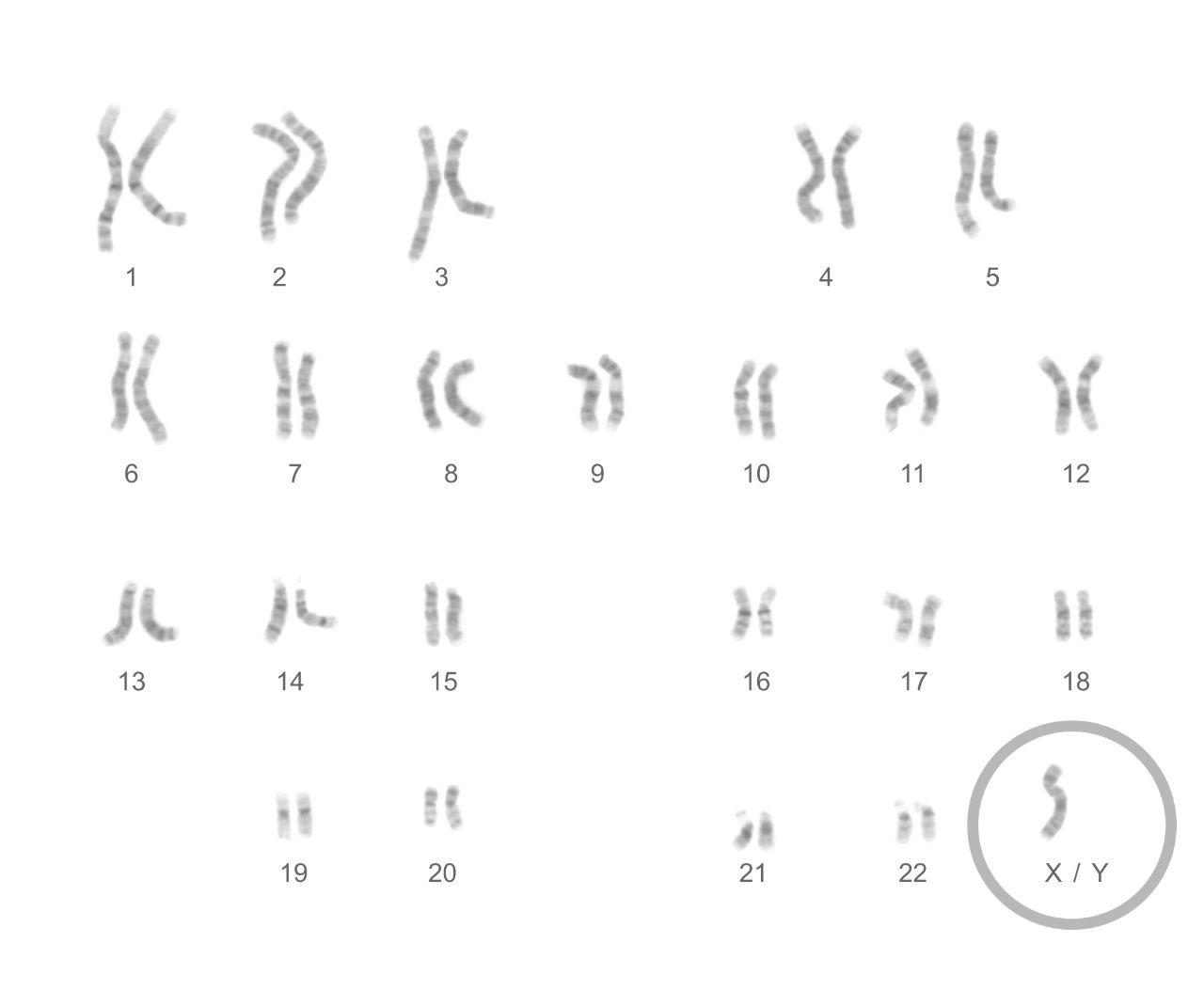 Turner syndrome females have a single X-chromosome and no other sex chromosomes.
Turner syndrome females have a single X-chromosome and no other sex chromosomes.
In addition to non-disjunction (leading to monosomies and trisomies), errors can also occur during crossing over (the exchange of materials between chromosomes that takes places prior to the chromosomes' separation, discussed in part two). The mechanism of crossing over, meiotic recombination, is highly complex and has long been the focus of intense research, but at a conceptual level it's easy to understand as the exchange of corresponding segments of DNA from two homologous chromosomes. When this process works correctly, it results in a shuffling of genes between two chromosomes. However, if the exchange is unequal, this can lead to duplication and/or deletion of a section of a chromosome.
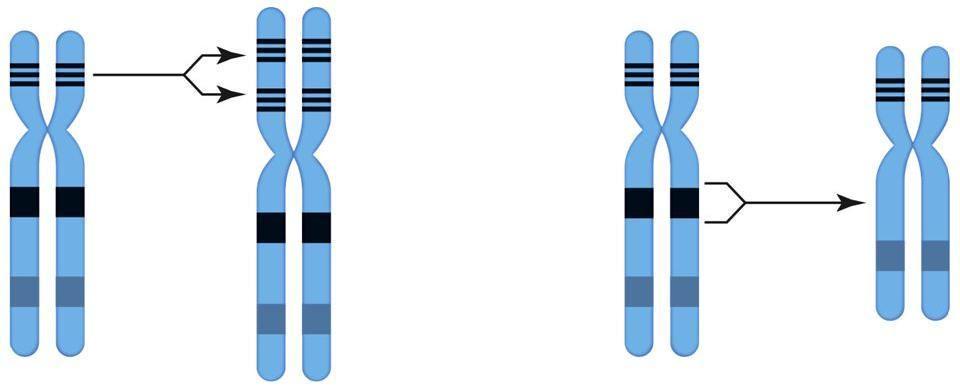 Left: duplication occurs when a segment of DNA is repeated, giving extra copies of the genes contained within the duplicated segment. Right: deletion occurs when a segment of DNA is removed from a chromosome, losing the genes contained within the deleted segment.
Left: duplication occurs when a segment of DNA is repeated, giving extra copies of the genes contained within the duplicated segment. Right: deletion occurs when a segment of DNA is removed from a chromosome, losing the genes contained within the deleted segment.
Duplications and deletions can vary greatly in size, incorporating few or many genes. Clearly, the larger the duplication or deletion, the more genes will be affected and the greater the effect on the organism carrying it. Also, as we have seen, in general duplication is less damaging than deletion.
Since crossing over relies on temporary breakage of chromosomes, there is also a risk that when the chromosome segments are rejoined, they can rejoin the wrong way around. This is called a chromosomal inversion, and although it usually doesn't affect the carrier of the inversion, it does lead to a reduction in fertility in proportion to the size of the inversion (when crossing over occurs within the inverted segment, the resulting chromosomes are unbalanced).
A more complex situation occurs when crossing over mistakenly occurs between two non-homologous chromosomes, leading to an exchange of DNA sequence between different chromosomes. Many of these translocations occur sporadically, but some specific ones recur in certain diseases, including many cancers (more on which later).
 Left: two different (non-homologous) chromosomes erroneously line up prior to crossing over. Center: a segment breaks off one member of each pair. Right: the non-homologous segments attach to different chromosomes. The resulting products are translocations.
Left: two different (non-homologous) chromosomes erroneously line up prior to crossing over. Center: a segment breaks off one member of each pair. Right: the non-homologous segments attach to different chromosomes. The resulting products are translocations.
Carriers of translocations usually have a balanced gene complement, but problems can occur when having children, the most obvious of which is reduced fertility. This reduction in fertility is due to the production of unbalanced gametes (i.e. gametes that have an extra segment of one chromosome and a deleted segment of another chromosome), which usually produce an unviable embryo following fertilization.
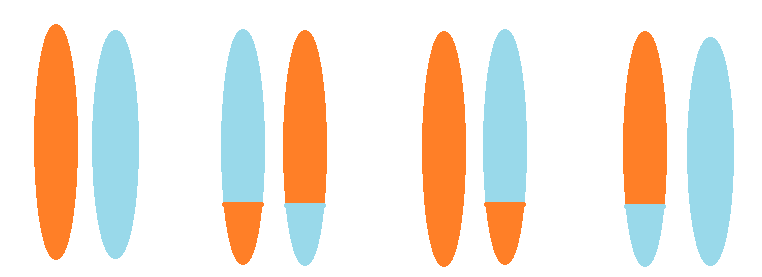 A carrier of a balanced translocation has one normal copy of each of two chromosome pairs, and one copy with a translocation. Since one of each homologous pair is inherited, there are four possible outcomes for offspring. Left: a normal copy of each chromosome. Left-center: a balanced translocation. Right-center: one chromosome is normal, but the other has a translocation; this is an unbalanced translocation. Right: also an unbalanced translocation, but the other way round.
A carrier of a balanced translocation has one normal copy of each of two chromosome pairs, and one copy with a translocation. Since one of each homologous pair is inherited, there are four possible outcomes for offspring. Left: a normal copy of each chromosome. Left-center: a balanced translocation. Right-center: one chromosome is normal, but the other has a translocation; this is an unbalanced translocation. Right: also an unbalanced translocation, but the other way round.
That completes our look at chromosomes. Let's now turn our attention back to the level of genes, and in particular to the different forms of these genes, the alleles. We've already considered dominant and recessive alleles and how they work, but now it's time to take a look at how these different alleles came to exist: the process of genetic mutation. Some of our genes will carry mutations that we inherited from one or both of our parents, which can lead to diseases that are passed down to later generations. In other cases, a new mutation occurs in a gamete, which leads to a disease in the offspring that was not present in either parent.
The A, C, G and T nucleotides that make up our DNA are functionally divided into groups of three consecutive nucleotides on the DNA strand. Since there are four different nucleotide letters, there are 43 = 64 possible combinations of 3-letter 'words', called codons, and each codon makes one specific amino acid out of a total of 20 (this redundancy means that most of the amino acids can be coded for by several different codons, as shown in the table below).
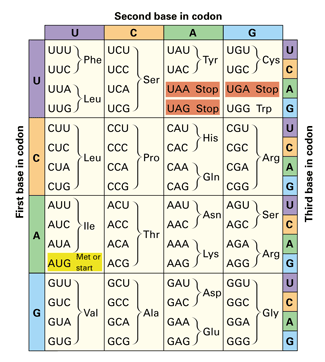 A table showing which 3-letter codons (upper case) code for which amino acids (three-letter abbreviations). Note the presence of start and stop codons. Note also that the nucleotides are labelled either A, C, G and U (rather than A, C, G and T). Don't let this confuse you; although A, C and G don't change between DNA and RNA, a letter T in DNA is equivalent to a U in RNA, and the table shows codons that have already been transcribed from DNA to RNA form.
A table showing which 3-letter codons (upper case) code for which amino acids (three-letter abbreviations). Note the presence of start and stop codons. Note also that the nucleotides are labelled either A, C, G and U (rather than A, C, G and T). Don't let this confuse you; although A, C and G don't change between DNA and RNA, a letter T in DNA is equivalent to a U in RNA, and the table shows codons that have already been transcribed from DNA to RNA form.
Remember that most nucleotides within a strand of DNA are functionless, so cells need a way to find the islands of genes in the much larger empty sea surrounding them. This is accomplished via 'start' and 'stop' codons: one particular codon is used to indicate the start of a gene and another (actually, one of three variations) is used to indicate the end.3
Because each specific three-letter codon codes for a specific amino acid, if there is a mutation (if one of the nucleotides is changed to a different nucleotide) this will often cause the codon to code for a different amino acid, and thus the resultant protein that is made will also often be different. The proteins that are built up from amino acids are very complex macromolecules that each form via an intricate and convoluted folding process; protein folding is highly sensitive to changes in amino acid sequence.
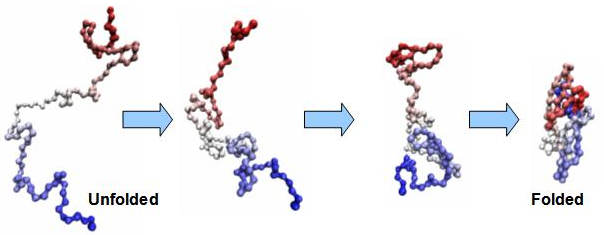 Protein folding. On the left is a sequence of connected amino acids, formed from the translation of RNA into protein. This sequence undergoes a complex biochemical folding process, based on the specific properties of each amino acid in the sequence. This results in a final folded protein of specific shape and function. Any changes to the amino acid sequence will usually affect the folding process and thus the final protein product.
Protein folding. On the left is a sequence of connected amino acids, formed from the translation of RNA into protein. This sequence undergoes a complex biochemical folding process, based on the specific properties of each amino acid in the sequence. This results in a final folded protein of specific shape and function. Any changes to the amino acid sequence will usually affect the folding process and thus the final protein product.
Each of the 20 amino acids have differing chemical properties, so if one amino acid is replaced by another in a sequence, this can have a dramatic effect upon folding and thus on the final protein produced. This type of mutation is called a missense or point mutation, since the mutation occurs in a single nucleotide and leads to the production of one different amino acid in the protein. The effects of this can be large or small, depending on the specific amino acid change and its location in the protein.
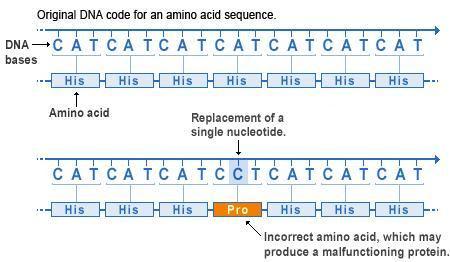 Missense mutations. At the top we see how the nucleotide sequence of a gene forms 3-letter codons, each coding for a specific amino acid. The amino acids are collectively aligned into a protein of a given sequence. Below we see a point mutation in the fourth codon has changed the original letter A to a letter C, resulting in a change of codon from CAT to CCT, which in turn changes the amino acid from His to Pro. Missense mutations like this will often affect protein folding and function.
Missense mutations. At the top we see how the nucleotide sequence of a gene forms 3-letter codons, each coding for a specific amino acid. The amino acids are collectively aligned into a protein of a given sequence. Below we see a point mutation in the fourth codon has changed the original letter A to a letter C, resulting in a change of codon from CAT to CCT, which in turn changes the amino acid from His to Pro. Missense mutations like this will often affect protein folding and function.
Another type of mutation whose effects are always large is called a nonsense or stop mutation. This occurs when the mutation causes the codon to change from coding for one of the twenty amino acids to coding for a 'stop' (one of the 3 out of 64 codons that indicates the end of a gene sequence). This type of mutation prematurely terminates the transcription of the RNA sequence. As this usually occurs in the interior of a sequence, it results in the loss of many amino acids; a nonsense mutation therefore tends to destroy the entire function of a protein.
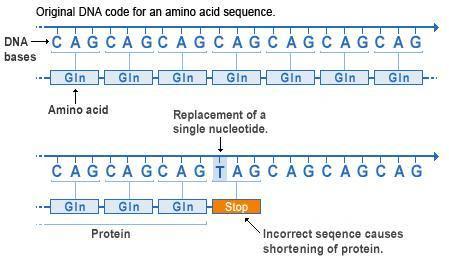 Nonsense mutations. Again we see an original sequence of codons at the top, with the same sequence following a point mutation below. However, in this case the codon is changed from CAG to TAG. From the table above we can see that the DNA codon TAG (transcribed to UAG in RNA) is a stop codon. So therefore the codon sequence is terminated at this point, and the remaining codons are not transcribed into RNA or translated into amino acids. This drastically truncates the protein, nearly always destroying its function.
Nonsense mutations. Again we see an original sequence of codons at the top, with the same sequence following a point mutation below. However, in this case the codon is changed from CAG to TAG. From the table above we can see that the DNA codon TAG (transcribed to UAG in RNA) is a stop codon. So therefore the codon sequence is terminated at this point, and the remaining codons are not transcribed into RNA or translated into amino acids. This drastically truncates the protein, nearly always destroying its function.
If we consider the genome as a whole, most mutations are actually irrelevant, as most of the genome is insensitive to sequence. In sections of the genome that are sequence-sensitive (such as those sections that are part of genes), most mutations are harmful, and most of the rest are neutral or nearly neutral. That most mutations are harmful should be intuitively obvious from the aphorism if something isn't broken, don't try to fix it; any changes made to a functional object are most likely to break the object. Most of the ways to change an object that don't break it still won't improve it (this corresponds to neutral genetic changes). The few remaining changes are beneficial.4 It is these neutral or beneficial mutations that occasionally become established in a population as a new allele for a specific gene.
Genetic mutations have several causes: errors during DNA replication leading to the wrong nucleotide being placed into a daugher sequence; environmental factors (e.g. many chemicals and ultraviolet light) that damage DNA which is subsequently affected by errors during repair; or from the insertion or deletion of DNA segments (called transposons) that can move around the genome.
Mutations are the raw material of evolution as well as a cause of many diseases, but as we aren't going to discuss evolution in this series, here I'm just going to focus on a certain type of harmful mutation: those that can cause cancer.
Cancer is a genetic disease. Many people carry known cancer risk mutations and can pass these on to their children. All of us acquire genetic damage during the course of our lives, some of which is carcinogenic. Virtually everyone who lives to later adulthood develops multiple cancerous or precancerous cell clusters (tumours) in various parts of their bodies.
Cancer is simply uncontrolled cell division. You will remember from earlier in this series that as old cells age, they wear out and must be replaced. Cells replace themselves by dividing, which they do once per cell cycle. This process of cell division is normally tightly controlled; a cell must only be allowed to divide when appropriate, so most of the time the cell cycle is paused at one of several cell-cycle checkpoints.
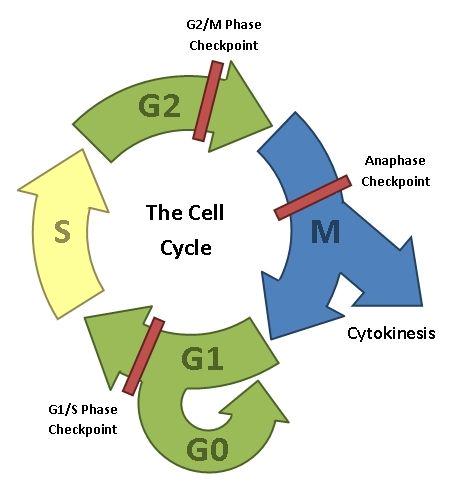 The cell cycle. After a cell has divided, the daughter cell is either in G1, where the non-genetic cell contents are duplicated ready for the next cell division, or it exits the cell cycle by going into a non-duplicative state called G0. In S-phase, the chromosomes are duplicated ready for the next cell division. The cell performs quality checks on the duplicated genetic material in G2, making any needed repairs. Mitosis and cytokinesis are the actual processes of cell division (discussed previously). Note that G1, G2 and Mitosis all have checkpoints associated with them, which halt the cell cycle until various necessary processes have been carried out.
The cell cycle. After a cell has divided, the daughter cell is either in G1, where the non-genetic cell contents are duplicated ready for the next cell division, or it exits the cell cycle by going into a non-duplicative state called G0. In S-phase, the chromosomes are duplicated ready for the next cell division. The cell performs quality checks on the duplicated genetic material in G2, making any needed repairs. Mitosis and cytokinesis are the actual processes of cell division (discussed previously). Note that G1, G2 and Mitosis all have checkpoints associated with them, which halt the cell cycle until various necessary processes have been carried out.
This regulation of the cell cycle is controlled by genes called cell-cycle inhibitors, the most well-known of which is a tumour suppressor called p53. This protein works to repair damaged (mutated) DNA, and can also pause the cell cycle to allow damaged cells to be repaired or, failing this, initiate cell death in unrepairable cells.
Like most genes, the one that codes for p53 is part of a genetic pathway, in which it interacts with other genes via proteins and RNA molecules. If the gene for p53 gets damaged, the entire pathway of which it is a part is affected, and p53s vital functions as a tumour suppressor are eliminated, which makes the development of continuous, uncontrolled cell division much more likely. Most human tumours contain a mutation of the p53 gene.
Usually, multiple carcinogenic mutations are required before an individual will go on to develop cancer. Luck plays a big role in this, but of course we'd also like to minimize mutation rates as much as possible, and so reducing exposure to known carcinogens can go a long way in preventing cancer.
A common approach to cancer treatment is specifically to attack those cells undergoing uncontrolled cell division (i.e., tumour cells). The two usual methods of doing this are chemotherapy and radiotherapy. Tumour cells can be specifically targeted precisely because they are undergoing rapid cell division; recall that cell division requires the genome to be copied, and for this to occur the DNA must unwind to allow access by the replicative protein machinery. In this state the DNA is most exposed to damage, whether via chemicals or radiation.
But mutations aren't always bad; we can also make them work for us, even if we don't cause the mutations ourselves. One example of this is selective breeding, which we have been doing for millennia. Selective breeding is a way to get organisms to have particular traits that we find agreeable. All domesticated animals as well as all our food crops have undergone extensive selective breeding. For example, all modern dog breeds were developed by selecting for mating just those animals with the desired traits for the particular breed standard being aimed at. Every dog currently existing is descended over thousands of years from a species of wild wolf, and is a cousin of modern grey wolves.
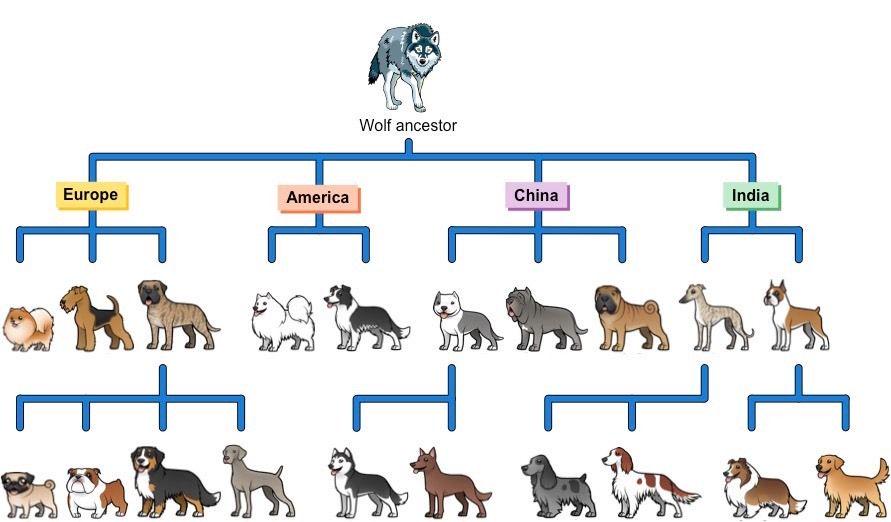 All dog breeds descend from wild wolf ancestors. Traits such as docility, loyalty, and tameness were selectively bred into the lineages over many generations.
All dog breeds descend from wild wolf ancestors. Traits such as docility, loyalty, and tameness were selectively bred into the lineages over many generations.
Plants have also been selectively bred for thousands of years. Originally, wild plants were cultivated and those specimens with the best traits (yield, and drought-, insect- and disease-resistance) were picked for reproduction whilst less desirable specimens were eliminated by preventing them from reproducing.
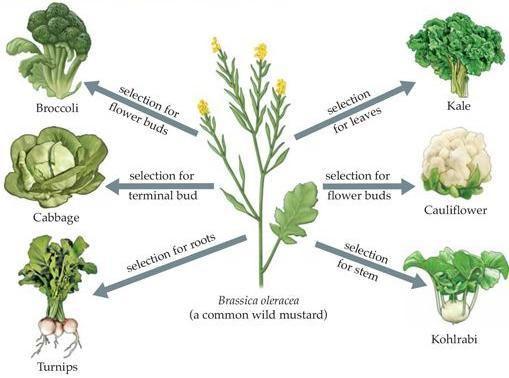 Broccoli, cabbage, turnip, kale and cauliflower were all bred from a particular species of wild mustard. The various traits of each plant type were selectively bred for, as shown.
Broccoli, cabbage, turnip, kale and cauliflower were all bred from a particular species of wild mustard. The various traits of each plant type were selectively bred for, as shown.
Selective breeding can take dozens of generations to fully incorporate desired characteristics. It's also prone to various problems, such as inbreeding. Inbreeding results from the repeated breeding of closely-related individuals over several generations, and leads to increasing loss of genetic variability as each generation goes by. This makes organisms more susceptible to various diseases, due to harmful recessive mutations getting fixed in a population, and also leaves the species as a whole less able to adapt to new environmental challenges that arise.
In recent years, geneticists have developed the ability to directly modify specific genes, which greatly speeds up the process of improving species of interest, as well as potentially avoiding the inbreeding problems associated with traditional selective breeding. Genetically modifying organisms in this way remains controversial, not least because of the abruptness of the change, and also because genes are frequently transferred across species. The ethical implications of this need not concern us here.
Genetics is now a high-technology discipline, and research is intense in many fields. In a future article, I'll give you an overview of some of this current work, along with informed speculation on some of the directions in which the field may go over the coming years.
Let's now conclude this series on genetics with a case study, bringing together many of the concepts we've covered over the three articles.
1 out of every 2 million people are unlucky enough to be born with a genetic mutation that leads to a condition called fibrodysplasia ossificans progressiva (FOP). As with several rare bone-related disorders, this is a particularly nasty disease, characterized by the progressive replacement of connective tissue (muscles, tendons and ligaments) by bone tissue. This process starts with the neck and shoulders, working its way down the body. Subsequent injuries and infections accelerate disease progression, often leading to the complete seizing-up of joints and a gradual and irreversible loss of mobility.
FOP is an autosomal dominant disease caused by a sporadically recurring mutation in a gene called ACVR1, which is located on chromosome 2. This mutation usually occurs de novo (i.e., it originates in the foetus, rather than being inherited from a parent), and leads to a change in a single amino acid. FOP is usually5 caused by a mutation in codon 206 of ACVR1, where a change from G to A leads to the production of the amino acid histidine in place of the similar amino acid arginine. This simple event, occurring in one parent's gamete and thus being present in all the cells of the offspring's body, is all that is necessary to cause FOP and the many severe symptoms of the disease.
There is a specific class of protein that receives chemical signals originating outside the cell, called protein receptors, which respond to these signals by modifying some activity of the cell. ACVR1 is part of a family of protein receptors known as bone morphogeneic proteins, type 1 (BMP1). BMPs are cellular growth factors, and the BMP1 family are involved in bone and cartilage development. ACVR1 codes for a protein that is located in the body cells' plasma membrane and acts as a signal transducer, facilitating the transfer of information between the inside and outside of the body's cells.
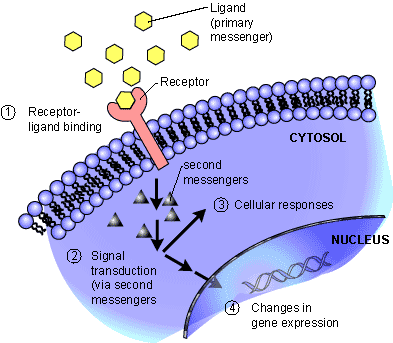 Signal transducers, such as the protein coded for by the ACVR1 gene, are located in the plasma membrane of the cell. They can transfer signals from outside the cell to inside the cell via binding with messenger molecules (ligands). They then pass the signal into the cell using internal messengers, which trigger a chain reaction that eventually results in changes in gene expression.
Signal transducers, such as the protein coded for by the ACVR1 gene, are located in the plasma membrane of the cell. They can transfer signals from outside the cell to inside the cell via binding with messenger molecules (ligands). They then pass the signal into the cell using internal messengers, which trigger a chain reaction that eventually results in changes in gene expression.
The mutation in ACVR1 is of a type known to geneticists as gain-of-function: a class of mutations that result in the body producing a protein that has an abnormal function. In the case of FOP, the amino acid change results in the ACVR1 protein having a modified physical shape. Normally, protein receptors are inactive until they are activated by another specific protein binding to them (see above figure). However, the modified shape of the mutated ACVR1 protein leads to the receptor becoming active without requiring interaction with its normal binding partner. This increases its sensitivity, meaning that the gene is continuously activated at a low level, and is also much more prone to being fully expressed at inappropriate times.
Although the ACVR1 mutation must be present to cause FOP, it seems that this alone is not sufficient to cause the disease. Geneticists use the concept of expressivity to describe observed variability in the type, number and severity of the symptoms associated with a disease caused by a specific mutation. FOP exhibits variable expressivity: fairly large differences in the frequency and severity of flare-ups and disease progression between patients. Environmental circumstances (for example bodily injuries and infections) clearly play a role, but secondary mutations in the same molecular pathway could also have an effect, as could differences in immune system function and in the immediate environment of the soft tissue that is affected. These are questions that researchers are actively working on.
That concludes this three-part introduction to genetics. In future articles, I'll build on this background knowledge to discuss more specific aspects of genetics and how these relate to biology as a whole.
Chromosome 21 has the fewest genes of all the autosomes, making trisomy 21 the mildest of the trisomies. The next two chromosomes with the fewest genes are 18 and 13. Trisomy 13 (Patau syndrome) and trisomy 18 (Edwards syndrome) foetuses can survive to birth, but do not normally live more than a few months at most. Trisomies of other autosomes are lethal in utero.
Although we learned in part two that most genes are haplosufficient (a single functional copy is enough to produce the required amount of protein), a significant fraction of genes are haplo-insufficient (and so require both copies to be present and functional). As even the smallest chromosomes have hundreds of genes, this means that a monosomy will affect scores of genes that are haploinsufficient.
Actually, the genes themselves are divided into alternating sections called introns and exons. The exons are collectively spliced together when building an RNA strand representing an entire gene, while the introns are discarded. Exons can be spliced together in different combinations, so that a single gene can actually code for several different proteins.
At some future date I intend to write several articles on evolution, the unifying principle of all of biology. It's enough to say here that those few mutations that turn out to be beneficial have done a lot of the heavy lifting in building organisms over huge time spans.
There are a few reported cases where a different mutation in the same gene has occurred.